a review on protein language models
Protein “language” is a lot like human language. Given the similarities, researchers have been building and training language models on protein sequence data, replicating the success seen in other domains, with profound implications. In this post, I will explore how transformer models have been applied to protein data and what we’ve found.
The language of proteins
Much like words form sentences, protein sequences—strings of the 20 amino acids that make up the protein "vocabulary"—determine the structure and function of proteins in their environment. This ordering of amino acids is crucial, as it influences how proteins fold and interact within biological systems.
Like human languages, which utilize modular elements like words, phrases, and sentences, proteins consist of motifs and domains. These are the fundamental building blocks reused in various combinations to construct complex structures. In this analogy, protein motifs and domains are akin to the 'words' and 'phrases' of the protein world, with their biological functions similar to the 'meaning' conveyed by sentences in human languages.
Another key parallel to human language is the concept of information completeness. Although a protein is much more than its amino acid sequence—it forms structures that carry out specific functions—all these aspects are predetermined by its sequence. Even though the way a protein behaves can vary depending on its environment and interactions with other molecules (e.g. on cellular state, other molecules and post-translational modifications), it is still defined by the underlying sequence. This means that from an information-theory perspective, the protein’s information (e.g. its structure) is contained within its sequence.
[Its important to keep in mind that the analogies between NLP and protein language modeling only go so far. First, we can read and understand natural language, and not so much with proteins. Unlike proteins, most human languages include uniform punctuation and stop words, with clearly separable structures. And while natural languages have a well-defined vocabulary (with ~ million words in English), proteins lack a clear vocabulary. Moreover, with proteins we do not always know whether a sequence of amino acids is part of a functional unit (e.g. a domain). Additionally, proteins also exhibit high variability in length ranging from 20 to a few thousand AA.]
Protein language model architectures
Encoder models
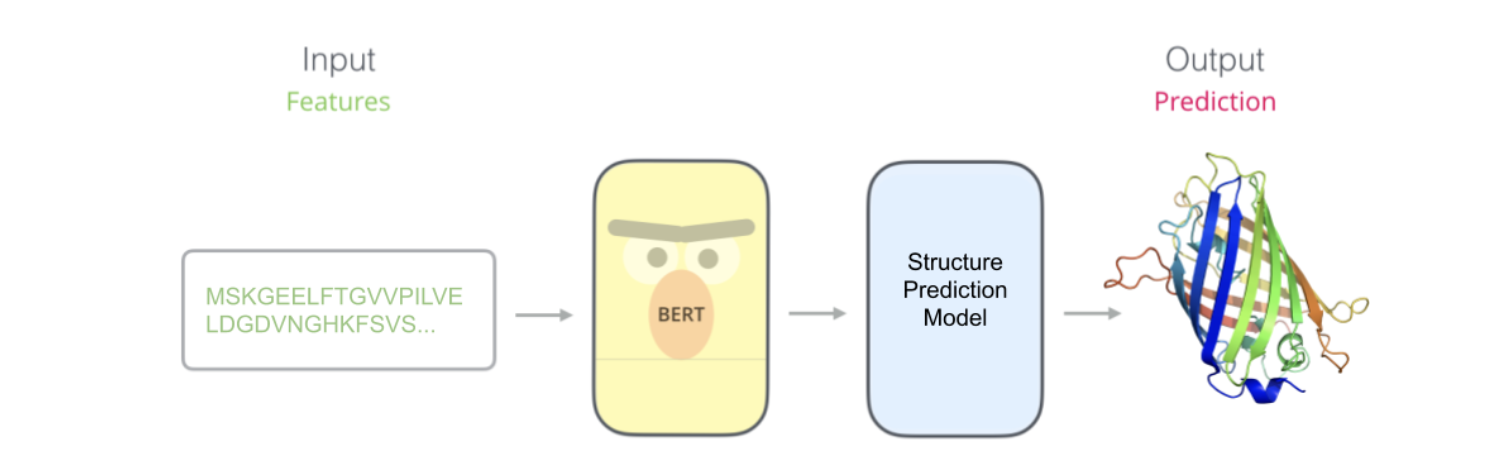
A lot of the initial work in pLMs is based on Encoder-only Transformer architecture as their aim is to obtain embedded representations of proteins in a vector space for downstream tasks.
Most of these models use BERT-like architectures and denoising autoencoding training objectives, i.e., they are pre-trained by corrupting input tokens in some way and trying to reconstruct the original sequence. Examples of such models include TCR-BERT, epiBERTope, ESM, ProtTrans or ProteinBERT. After pre-training to generate embeddings, these models are further refined through supervised learning techniques to address various downstream tasks in protein engineering (secondary structure and contact prediction, remote homology detection, post-translation and biophysical properties prediction)
Decoder models
Unlike encoder models, decoder models adopt autoregressive training, a method where models are trained to predict subsequent words based on a given context. The most well-known of these types of models are, you guessed it, the GPT-x models.
An early example of the GPT style decoder models in the protein world is ProtGPT2, trained on 50M sequences with 738M parameters.
Did it work?
Kind of. ProtGPT2 managed to generate sequences with characteristics similar to those of natural proteins. The amino acid types and frequencies closely matched those in nature, and the sequences exhibited a comparable balance between ordered (stable) and disordered (flexible) regions. And visual inspection of the structural superimposition showed that generated proteins preserved some of the binding sites, which is essential for functionality. So, although the generated sequences looked a lot of natural proteins, it's hard to say if they really functioned like one. It was quickly outpaced by bigger and better models.
Conditional transformers
While ProGPT2 leverages a GPT2-like architecture for general protein sequence generation, newer approaches have been developed that integrate deeper biological contexts during the training phase. These methods ensure the patterns learned are not only statistically correct but also biologically meaningful. Protein models can be conditioned in two main ways: i) by conditioning on sequences, or ii) by conditioning on the structure of the proteins.
Conditioning on sequence:
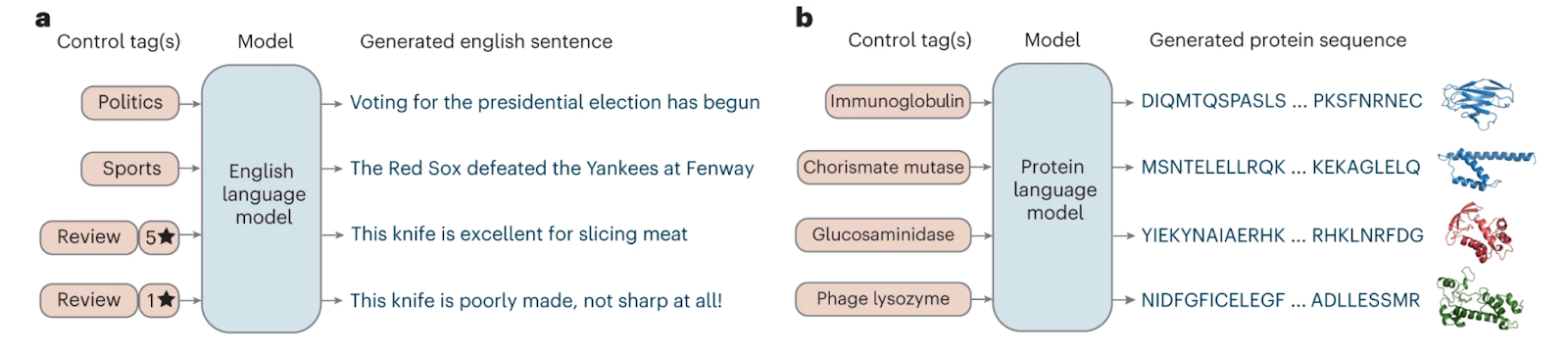
The development of Conditional TRansformer Language (CTRL), an autoregressive model that includes conditional tags, marked a significant advance in NLP. These tags allow for targeted text generation without requiring input sequences. Known as control codes, these tags significantly refine the influence over genre, topic, or style, marking a major advance toward targeted text generation.
So naturally, CTRL was soon adapted to a dataset of 281 million protein sequences. The resulting model, named ProGen, employs UniProtKB Keywords as conditional tags. These tags span ten categories such as ‘biological process’, ‘cellular component’, and ‘molecular function’, encompassing over 1,100 terms. ProGen achieved perplexities on par with high-quality English language models, even when generating sequences for protein families not included in its training set.
ProGen's performance shows significant advances in our ability to design proteins that behave like natural ones. This model has successfully created protein sequences that function effectively, as proven by rigorous tests where they performed as well or better than naturally occurring proteins. Specifically, ProGen was able to:
- Create proteins that match the energy efficiency and structural accuracy of naturally occurring proteins.
- Generate variations of a specific protein domain that proved to be more effective than random variations, suggesting a high degree of predictive accuracy and utility in practical applications.
And now it can do so much more, as shown Profluent bio’s new paper where they used ProGen to design Cas9 protein, which didn’t exist in nature but was able to edit genes successfully in humans. These achievements are important because they indicate we can now design synthetic proteins that not only mimic but improve upon natural proteins. I expect more cool stuff like this in the near future.
Open questions:
- How well does ProGen scale to even larger datasets? Are there limits to the number of protein families or conditions it can effectively model?
Conditioning on structure:
Besides sequence, we can incorporate the structure of a protein while training such that the model can learn structure —> sequence. This is called “inverse folding” because it does the exact opposite of protein folding which is sequence —> structure.
That may sound counterintuitive, but this is actually an incredibly useful process for protein design, especially for enzymes and therapeutics. Let's say you have a specific task you want your enzyme or therapeutic protein to perform, such as binding to a certain molecule or catalyzing a particular reaction. Traditional methods often involve tweaking existing protein sequences and testing whether the new versions do the job better. This can be slow and somewhat hit-or-miss.
Inverse folding, on the other hand, starts with the ideal structure in mind — the one you predict will best perform the task. From there, it works backwards to figure out which sequences can fold into that structure. This approach has several advantages. For starters, inverse folding models tend to be very fast and can predict hundreds of sequences that might fold into a desired structure within minutes. It also explores a broader range of potential sequences than traditional methods, which might only alter existing sequences slightly. This broad exploration increases the chances of finding a sequence with optimal properties that hasn't been considered before.
ESM-IF is one example of an inverse folding models trained on AlphaFold Database (12M) and CATH Protein Structure Classification Database (~16,000) that uses an encoder-decoder architecture that takes structures as inputs to the encoder and autoregressively decodes sequences conditioned on structural encodings.
Open questions:
- Given that inverse folding models are being trained on AlphaDB, which contain predicted rather than experimentally determined structures, how can we effectively assess the reliability and accuracy of the protein sequences generated by inverse folding models?
Scale is all you need
So we’ve seen the performance on complex tasks improve in general-purpose language models as compute, data and model size increases. At certain scales, language models exhibit useful capabilities which emerge as a result of scaling a simple training process to large corpuses of data, like few-shot language translation, commonsense reasoning, and mathematical reasoning.
A similar idea exists for inference from sequences in biology. Because the structure and function of a protein constrain the mutations to its sequence are selected through evolution, it should also be possible to infer biological structure and function from sequence patterns, which would provide insight into some of the most foundational problems in biology.
This is exactly what ESM-2, a 15 billion parameter model built by Meta does.
By leveraging the internal representations of the language model(ESM-2), ESMFold generates structure predictions using only a single sequence as input, resulting in considerably faster structure prediction. ESMFold also produces more accurate atomic-level predictions than AlphaFold2 or RoseTTAFold when they are artificially given a single sequence as input, and obtains competitive performance to RoseTTAFold given full MSAs as input. Moreover, we find that ESMFold produces comparable predictions to state-of-the-art models for low perplexity sequences, and more generally that structure prediction accuracy correlates with language-model perplexity, indicating that when a language model better understands the sequence, it also better understands the structure.
and
Atomic resolution structure prediction can be projected from the representations of the ESM-2 language models … (and accuracy improves with the) scale of the language model.
This is pretty incredible! By scaling up the model size and dataset size, we can get rid of specific inductive biases (e.g., MSA) and generate structure predictions using only a single sequence as input.
Although ESM-2 is less accurate than AlphaFold today, it is an interestingly straightforward approach that can utilize an ever-expanding pool of diverse and unannotated protein sequence data:
| AlphaFold | ESMFold | |
|---|---|---|
| Parameters | ~100M | ~15B |
| Training data | ~200k structures, MSA | ~65M sequences |
| Loss | Several loss functions used | Masked amino acid |
To further illustrate the mindblowing capabilities of the ESM protein models, researchers conducted a remarkable experiment with highly optimized monoclonal antibodies, including antibodies targeting diseases like Ebola and COVID. They input the sequences of these antibodies into the ESM model, which then identified discrepancies between the actual amino acid sequences and their predictions. By selectively substituting the amino acids at these divergent sites with those predicted by the model, the researchers significantly enhanced the binding affinity, thermostability, and in vitro potency of the antibodies—achieving increases of up to sevenfold for mature antibodies and an astonishing 160-fold(!!) for unmatured ones.
Open questions:
- What are the limits of scaling laws for protein models, and at what volume of data do these models reach their maximum effectiveness?
Conclusion
Despite all the significant progress, we are still in the early stages of fully grasping the complexity of protein sequence space. For a long time, the only method to understand the relationship between proteins involved explicit pairwise or multiple sequence alignments, which rely on presumed evolutionary connections to map one protein's residues to another's. Recently, however, a more generalized approach is taking shape, focusing less on evolutionary lineage and more on the essential functional and structural aspects of proteins. If this pace of progress continues, we stand on the brink of potentially groundbreaking discoveries—uncovering unknown facets of familiar proteins and even synthesizing entirely novel proteins.
One challenge to this vision is the (lack of) human interpretability of such representations. As these models become more complex, understanding how they process and represent protein sequences is crucial, especially for applications like drug discovery where identifying how models predict binding sites can be very useful. The next steps in protein modeling also involve developing more biologically inspired models. This means deepening our integration of biochemical knowledge within the frameworks of these models to refine their accuracy and functionality. By embedding more profound biological insights into model training and data processing, we can significantly improve outcomes across various protein-related tasks.
It was a lot of fun diving into protein language models. As I read more, the potential for more breakthroughs in protein science seems promising, especially by combining bigger and better models with clever experiment design. I’m excited to see what the next steps in protein modeling bring us!
Citation
Cited as:
Srinivasan, Apoorva. (May 2024). A Review on Protein Language Models. Apoorva Srinivasan's Blog. https://www.apoorva-srinivasan.com/plms/